Estudio de asociación de genoma completo (GWAS) para el análisis de la microbiota rizosférica
Aunque se sabe que el genotipo de las plantas influye en la microbiota con la que pueden interactuar, la manera en que los genes específicos dirigen las interacciones entre las plantas y sus hospederos es un tema que no ha sido estudiado a profundidad. Esto se debe a que algunas características ligadas a las plantas como como el perfil de metabolitos exudados o la arquitectura de sus raíces están determinadas por un gran número de genes. En éste sentido, los estudios de asociación de genoma completo (GWAS por sus siglas en inglés), permiten identifcar microbios sensibles al genotipo del hospedero y ligarlos con loci que puedan estar relacionados con su colonización.
Para estudiar la relación entre el genotipo y la microbiota, los autores de éste trabajo utilizaron plantas de sorgo de una colección de germoplasma que ha sido previamente genotipificado y analizaron la microbiota de hojas, rizósfera y endósfera de raíz por medio de la secuenciación de amplicones de la región V3-V4 del gen 16S rRNA de 24 genotipos distintos. Al relacionar las diferencias genotípicas, con las diferencias en la microbiota por medio de una prueba de Mantel, sólo se encontraron diferencias significativas en las muestra de rizósfera, por lo que se analizó la diversidad microbiana de rizósfera de 200 genotipos distintos de sorgo.

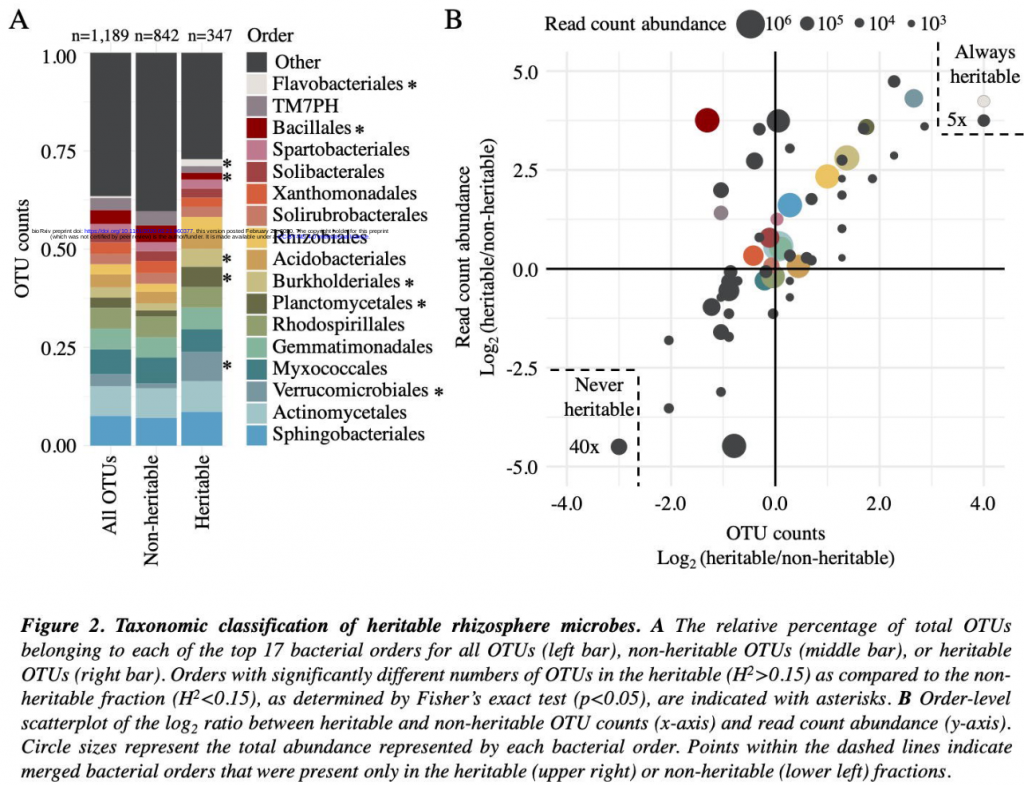
Al estimar la heredabilidad los OTUs y dividirlos como heredables (heredabilidad mayor a 0.15) o no heredables (heredabilidad menor a 0.15), encontraron perfiles filogenéticos distintos en cada grupos de OTUs. De manera general las clases que presentan cambios de abundancia entre heredables y no heredables incluyen a Flavobacteriales y Bacillales. Los 6 Flavobacteriales detectados solo se encuentran en la fracción heredable; mientras que los Bacillales presentaron un bajo número de OTUs heredables, pero cuyos lecturas fueron encontrados en alta abundancia, lo que sugiere su elevada abundancia en la rizósfera.
Para realizar el GWAS se emplearon tanto las propiedades globales de las comunidades microbianas (componentes principales en el análisis de ordenamiento), como las abundancias de los OTUs individuales. El GWAs realizado con el componente principal 1 (PC1), que explica el 21% de la varianza, reveló una correlación significativa con un locus de aproximadamente 1.15 Mb del cromosoma 4 y un umbral riguroso. El GWAS basado en la abundancia de cada OTU, permitió encontrar dos grupos de OTUs distintos (de 39 y 10 OTUs respectivamente) con correlaciones significativas como los cromosomas 4 y 6. Al realizar un análisis de SNPs del locus detectado en el cromosoma 4 reveló la existencia de dos alelos, un alelo mayor (presente en la mayoría de los genotipos y un alelo menor (presente en la minoría). La mayoría de los OTUs presente en el alelo mayor pertecen a bacterias monodermas y las del alelo menor, pertenecen principalmente a bacterias didermas, lo que les sugierió que los mecanismos genéticos del hospedero interactuaban con carcaterísticas de las bacterias. Para explorar cuales eran los mecanismos genéticos que podrían estar implicados en la interacción, los autores del trabajo analizaron datos obtenidos por estudios previos de los patrones de expresión tejido específicos de los genes dentro del intervalo de 1.15 Mb del cromosoma 4. Los genes candidato con una mayor expresión en los tejidos de raíz incluyen una xilanasa y una anhidrasa carbónica, que podrían participar en la liberación de compuestos hacia la rizósfera.

Finalmente, con la finalidad de validar que la variación alélica del locus candidato en el cromosoma 4, se realizó una segunda caracterización del microbioma de rizósfera incluyendo genotipos de sorgo no contemplados con anterioridad, utilizando 50% de individuos con el alelo mayor y el resto con el alelo menor. Al realizar un análisis de ordenamiento se encontraron diferencias entre las muestras relacionadas con el alelo al que pertenecían, las cuales fueron verificadas mediante un análisis de PERMANOVA. Como se había visto con anterioridad, los OTUs asociados al aleo mayor pertenecían a linajes de bacterias monodermos y los del alelo menor a bacterias didermas.

Se espera que con éste tipo de estudios se logre una mayor compresión de los mecanismos particulares que median la interacción planta-microorganismo, así como promover el reclutamiento de microorganismos benéficos que mejoren el rendimiento de los cultivos.
Referencia:
Deng, S., Caddell, D., Yang, J., Dahlen, L., Washington, L., & Coleman-Derr, D. (2020). Genome wide association study reveals plant loci controlling heritability of the rhizosphere microbiome. BioRxiv.



