Comparando metagenomas con base en la composición nucleotídica
El análisis de datos metagenómicos se basa comúnmente en la reconstrucción de fragmentos genómicos, predicción de genes y su comparación con secuencias contenidas en las bases de datos. Sin embargo, tanto el ensamblado como la búsqueda contra secuencias conocidas (anotación) es un procedimiento lento y computacionalmente costoso. En este trabajo, se propone la aplicación del perfil de k-meros, o palabras de nucleótidos de cierto tamaño k, contenido en las secuencias metagenómicas como medida de diversidad beta para la comparación de comunidades microbianas.
Los autores comparan esta estrategia (BC k-mer) con métodos de metagenómica comparativa: mapeos taxonómicos a nivel de género y especie (BC TAX genus y org), el cálculo de una versión modificada de UniFrac para datos genómicos (WG UniFrac), búsqueda de marcadores moleculares específicos (BC MetaPhlAn) y anotación funcional clasificada por grupos de ortólogos (BC COG). Los datos analizados incluyeron metagenomas simulados y 353 metagenomas intestinales reales de población china (China) y estadounidense (HMP).
Se encontró que este método es robusto ante los errores de secuenciación y mutaciones genómicas. Además, esta medida fue capaz de delinear muestras con un amplio espectro de características funcionales. Esta aproximación resultó correlacionada en un mayor grado a la anotación funcional en comparación con la anotación taxonómica, probablemente por la variación en el contenido genético de las muestras. El análisis de k-meros en secuencias metagenómicas genera grandes cantidades de información de una manera computacionalmente eficiente y aprovecha toda la información contenida en las lecturas metagenómicas.
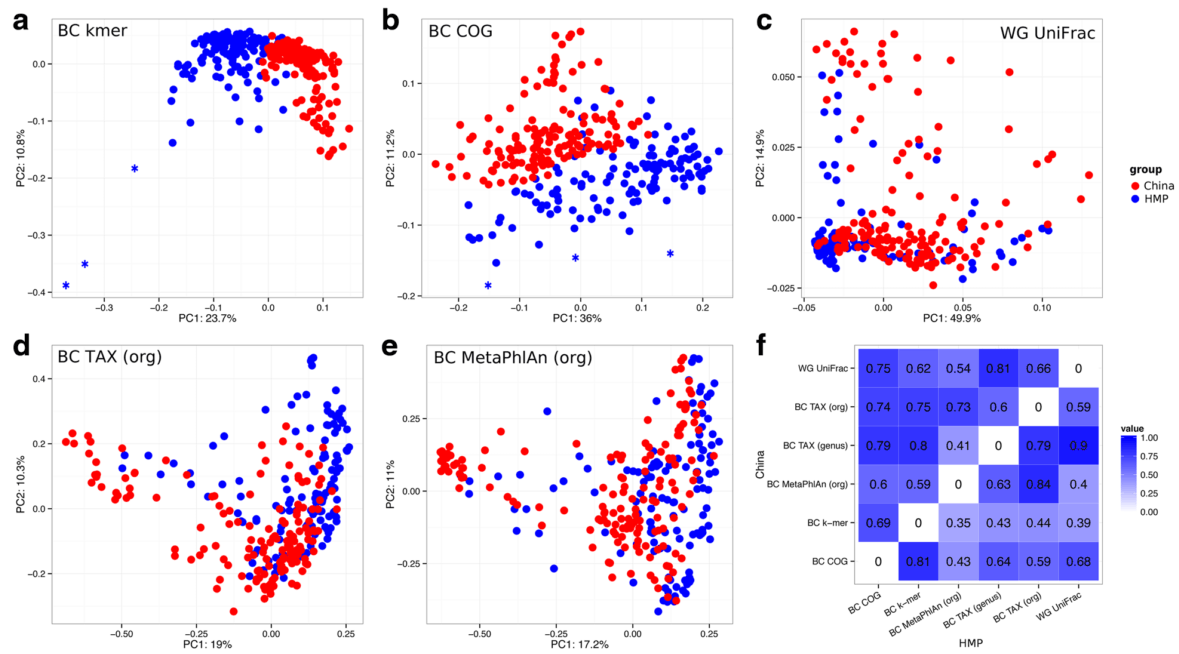
Variation of metagenomes using different dissimilarity measures. PCoA plots for different dissimilarity measures: a BC k-mer, b BC COG, c WG UniFrac, d BC TAX (org), e BC MetaPhlAn (org). Three samples-outliers are marked with asterisks. f Heatmap of Spearman correlation coefficient between dissimilarity matrices obtained using different measures (the upper triangle ofmatrix represents coefficients for China, the lower – for HMP)
Dubinkina, V. B., Ischenko, D. S., Ulyantsev, V. I., Tyakht, A. V., & Alexeev, D. G. (2016). Assessment of k-mer spectrum applicability for metagenomic dissimilarity analysis. BMC bioinformatics, 17(1), 38.


