Perfil de modelos ocultos de Markov para el reconocimiento de secuencias virales divergentes
Debido a que las tasas de mutación son relativamente altas para los virus, su reconocimiento ha resultado problemático y por ello se propone el análisis de secuencias virales como método complementario a técnicas de detección tradicionales. Para ello, en este trabajo se evaluó la capacidad de perfiles HMM virales (vFams) para clasificar con precisión las secuencias virales a partir de datos metagenómicos.
Es así que se generaron perfiles HMM a partir de secuencias de proteínas virales recuperadas del NCBI curadas en RefSeq. Estas secuencias se filtraron y se agruparon de acuerdo a diferentes criterios para posteriormente generar alineamientos múltiples de secuencias (ASM) significativos para construir perfiles HMM virales (vFams).
Las vFams se diseñaron con el objetivo de determinar si los conjuntos de datos metagenómicos contenían alguna secuencia viral, por lo que emplearon un experimento de validación cruzada ‘leave-one-out’’ para cumplir dos propósitos principales: determinar si podían reclutar proteínas homólogas desconocidas y para probar la capacidad vFam para distinguir con precisión la secuencia viral de la no viral.
Además de la longitud del perfil, el contenido de la información y el número de secuencias utilizadas, se incluyó la taxonomía de las secuencias a nivel de familia y género.
Los vFams fueron capaces de recordar al menos el 80% de sus secuencias excluidas y la variación en su capacidad de reconocimiento de secuencias virales también consideró la presencia de secuencias homólogas no virales; es decir, proteínas conservadas en eucariotas pero también presentes en los virus (inhibidores de apoptosis, metabolismo de nucleótidos, síntesis de DNA, replicación de DNA, modificación postraduccional de proteínas). Aunque estos vFams se eliminaron durante la etapa de filtrado, ya que su bajo recordatorio estricto y la falta de especificidad por las secuencias virales las hacen poco informativas y potencialmente engañosas.
Finalmente, para comparar el rendimiento de los perfiles HMM con BLAST se emplearon datos tres conjuntos de datos metagenómicos publicados previamente. El primer conjunto de datos procedía de muestras de diarrea de etiología desconocida, el segundo conjunto pertenecía a un aislado de un caracol en una uva podrida y el tercer conjunto de datos procedía de una biblioteca de secuencias de diferentes tejidos de una boa arborícola.
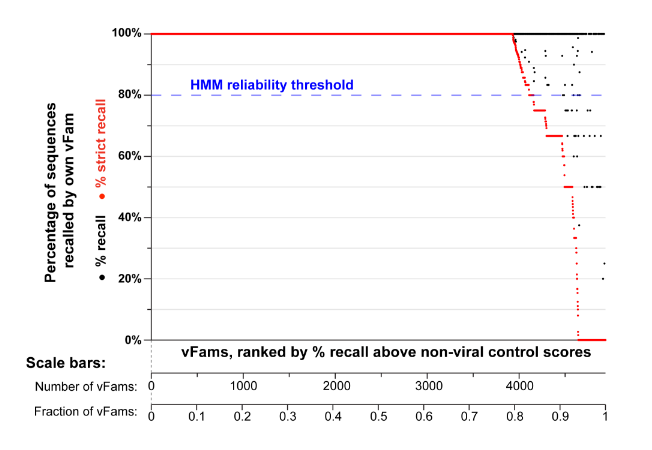
Figura 2. Si el perfil de HMM recupera la secuencia omitida con un E-value ≤10, la secuencia se considera «recordada». Si la validación HMM recupera la secuencia omitida y adicionalmente tiene un E-value más bajo que todas las secuencias de prueba, la secuencia se considera «recordada estrictamente».
De acuerdo a los experimentos de validación cruzada, se grafica el porcentaje de secuencias recordadas (negro) y el porcentaje de secuencias estrictamente recordadas . Las vFams se clasifican por su porcentaje de secuencias estrictamente recordadas (eje x). Se usó un umbral de 80% de recuperación estricta (línea azul discontinua) para filtrar las vFams.
Referencia:
- Skewes-Cox, P., Sharpton, T. J., Pollard, K. S., & DeRisi, J. L. (2014). Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PloS one, 9(8), e105067.