Megazords microbianos inversos o la descomplejización del microbiona del suelo
Los microoorganismos que habitan en los suelos desempeñan funciones importantes que incluyen la participación en el ciclaje de nutrientes y el establecimiento de interacciones que pueden favorecer el crecimiento de las plantas. Sin embargo, debido a la elevada complejidad de las comunidades que forman, los autores de éste trabajo buscaron estudiar sus capacidades metabólicas por medio de su división en módulos funcionales creados a partir de inóculos de suelo en medio M9, sometido a diversas condiciones. Dichas condiciones se pueden resumir en cinco categorías principales: sustratos simples (como azúcares y ácidos orgánicos), antibióticos, polisacáridos, anaeróbicos (con condiciones anoxicas y aceptores alternativos de electrones) y estrés (tres fuentes de carbono expuestas a diversas condiciones como como calor, bajo pH y alta salinidad). Con base en su diseño experimental, obtuvieron 324 comunidades comprendidas en 66 módulos funcionales distintos.
La diversidad de las comunidades fue analizada mediante la secuenciación masiva de amplicones del gen 16S rRNA. Al analizar la diversidad beta por medio de la métrica UniFrac ponderada, no encontraron algunas diferencias relacionadas con los módulos y la categoría a la que pertenecen. Por otro lado, resulta interesante que la mayoría de las clases del suelo nativo, así como su control (asilado de suelo en medio líquido) presentan clases de bacterias que se pierden en la mayoría de los tratamientos.

La pérdida de grupos bacterianos en los distintos módulos, se vió reflejada en la disminución generalizada de la riqueza y los índices de diversidad de Shannon y de Faith. Cabe resaltar que la diversidad encontrada en los módulos de polisacáridos fue mayor que en los sustratos simples, misma que también incrementó en algunos módulos estresados. Así mismo, se evaluó la reproducibilidad en los módulos por medio de la dispersión en la diversidad beta al interior de los módulos, observando una alta reproducibilidad en los sustratos simples y baja en otros módulos como el anaeróbico y el de polisacáridos. Sin embargo de manera general, los módulos son más similares entre sí, que respecto a otros módulos, indicando que hasta cierto punto, se puede replicar el experimento.

Para analizar las capacidades metabólicas de las comunidades seleccionadas, se analizaron los metatranscriptomas (por triplicado) de los módulos de n-acetilglucosamina, xilosa, gentamicina, xilano y pectina. Al realizar una prueba de Mantel para comparar la congruencia entre los datos de amplicones y de los metatranscriptomas se encontró correspondencia entre ambos tipos de datos. Además, como se observó al analizar la disperisón beta interna, los polisacáridos (xilano y pectina) mostraron myores diferencias que los sustratos simples (n-acetilglucosamina y xilosa). En el módulo de gentamicina, al analizar las secuencias obtenidas se observó un enrqiquecimiento en las categorías de aminoácidos, péptidos y aminas. Los ortólogos de KEGG en xilano y pectina, comprenden una mayor porción del mapa metabólico que aquellos enriquecidos en glucosa y n-acetilglocosamina, por lo que se infiere que los microbios de los módulos de polisacáridos tienen un mayor rango de expresión o la mayor riqueza taxonómica implica una mayor diversidad metabólica. Los transcritos más abundantes se relcionan con funciones de mantenimiento (como fosforilzación oxidativa y la síntesis de proteínas ribosomales).
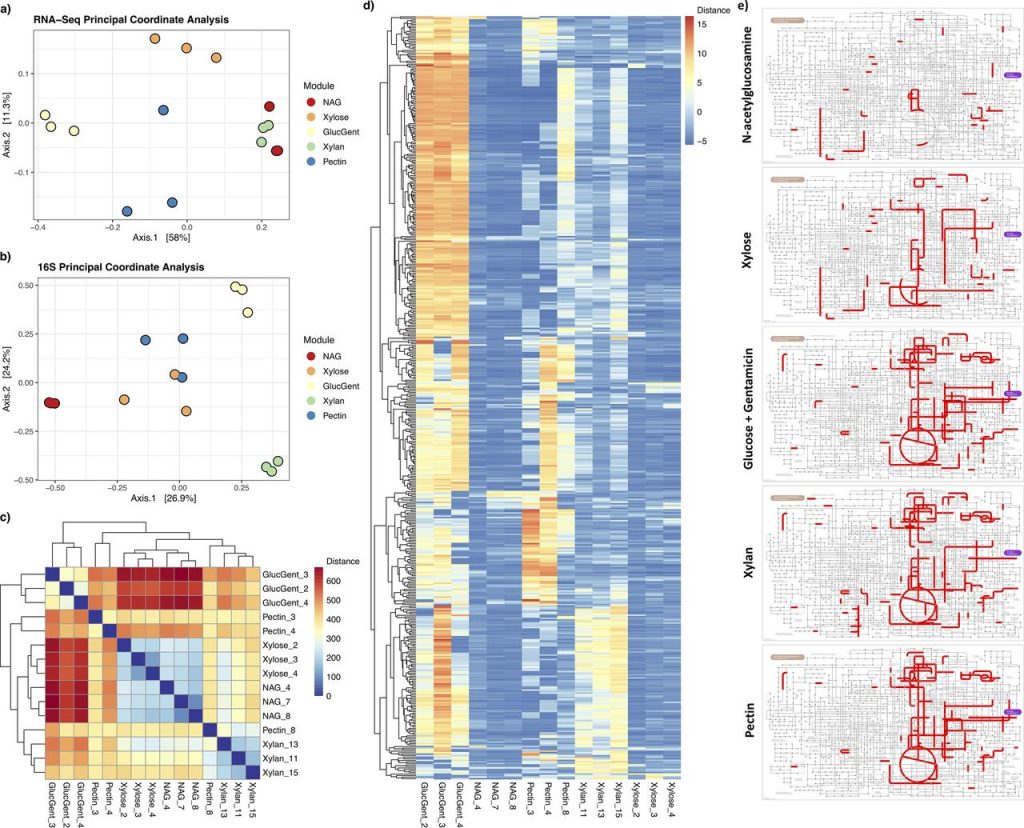
De éste modo, los autores consideran que es posible deconstruir la capacidad metabólica colectiva de de un microbioma complejo en componentes más pequeños con diferenctes perfiles de expresión, caracterizados por la pérdida de grupos taxonómicos y potencial metabólico. Resultando en módulos más pequeños que podrían ser estudiados con mayor facilidad.
Referencia:
Naylor, D., Fansler, S., Brislawn, C., Nelson, W. C., Hofmockel, K. S., Jansson, J. K., & McClure, R. (2020). Deconstructing the Soil Microbiome into Reduced-Complexity Functional Modules. Mbio, 11(4).