Los genes de virulencia de Helicobacter pylori tienen una alta estructura en las poblaciones americanas
Helicobacter pylori es una bacteria que habita el estómago humano que bajo ciertas condiciones puede generar úlceras y en casos graves cáncer. La demografía de esta bacteria refleja la del ser humano y ya que su transmisión se da por contacto cercano en familias y comunidades, se ha hipotetizado que existe una alta estructura en el bagaje genético de este organismo. Uno de los eventos de mezcla de poblaciones humanas más importantes fue la inmigración de individuos europeos al continente americano alrededor del siglo XVI. Aunque se sabe que este fenómeno tuvo un impacto en la diferenciación de las poblaciones de H. pylori en América, no está claro el efecto de esta migración en los genes de virulencia de este organismo como vacA, cagA y babA. Con el fin de conocer la estructura de H. pylori y sus genes de virulencia en el continente americano, Zilia Muñoz-Ramirez y compañía aislaron y secuenciaron organismos de esta especie en América y en la Península Ibérica y los compararon con los genomas conocidos de H- pylori. De esta forma, llevaron a cabo un análisis de 723 genomas provenientes de todo el mundo.
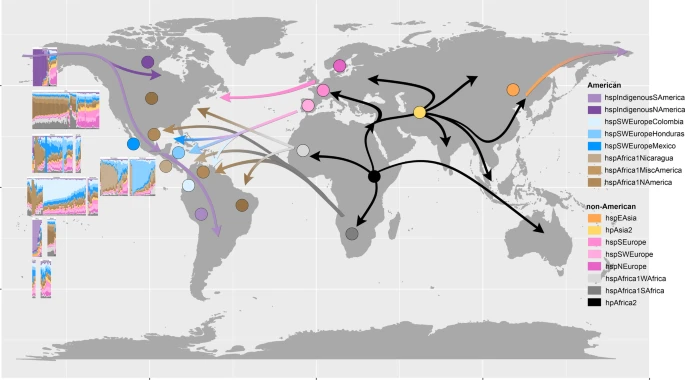
Al analizar la estructura de los genomas de H. pylori, los autores encontraron una alta contribución de genes de las poblaciones europeas en genomas de Colombia, Honduras y México. Sin embargo, las poblaciones con mayor carga europea fueron las de E.E.U.U., Canadá y Argentina. Otras asociaciones se encontraron entre genomas de E.E.U.U., Brasil, Nicaragua y Honduras con genomas del oeste de África. Curiosamente, la única población americana que se asoció con genomas asiáticos fue la de nativos americanos, que estuvo formada por dos poblaciones, una norteamericana y una sudamericana. Cuando buscaron el grado de mezcla en las poblaciones, encontraron que las poblaciones con menor grado fueron las nativas americanas y las de mayor grado fue la mexicana, probablemente por ser la puerta de entrada principal de los españoles en América.

De los 35 genes que mostraron alta estructura, 22 de ellos codifican para factores de virulencia y proteínas membranales, incluyendo vacA y babA. El gen con la estructuración más fuerte fue el que codifica para CagF, que es una chaperona de CagA. Los autores también encontraron mutaciones no sinónimas fijadas en dominios funcionales de las proteínas de virulencia. Dentro de esta diversidad genética, Muñoz-Ramirez y compañía encontraron patrones evolutivos distintos para los genes de interacción con el hospedero, que fueron a su vez distintos a los observados en el genoma núcleo de esta especie. En general, la distancia genética fue menor dentro de los genomas no americanos formando grupos aislados, mientras que los genomas americanos se encontraron dentro de grupos mezclados. Este trabajo nos ayuda a entender el porqué de la alta frecuencia de enfermedades asociadas a H. pylori en América y nos inspira a llevar a cabo análisis genómicos a gran escala.
Muñoz-Ramirez, Z.Y., Pascoe, B., Mendez-Tenorio, A. et al. A 500-year tale of co-evolution, adaptation, and virulence: Helicobacter pylori in the Americas. ISME J (2020). https://doi.org/10.1038/s41396-020-00758-0