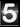El sesgo de uso de codones es útil para predecir tasas de crecimiento de comunidades microbianas
Conocer la tasa de crecimiento de los organismos es útil para llevar a cabo experimentos de ecología experimental y para modelar matemáticamente las dinámicas de crecimiento de comunidades. Gracias a la bonanza actual de datos de secuenciación, ha sido posible ligar ciertas características genómicas o de los propios datos de secuenciación a la velocidad de crecimiento de los microorganismos. Una característica genómica que se ha encontrado relacionada con la tasa de crecimiento es el sesgo de uso de codones (CUB), ya que se sabe que algunos codones se leen con mayor rapidez gracias a la disponibilidad diferencial de tRNAs. Por otra parte, la medición de la cobertura de secuenciación a lo largo del genoma tomando como referencia el origen de replicación (PTR) podría indicar la velocidad de crecimiento de una población de células. Aunque estas aproximaciones se han validado con cultivos bacterianos axénicos, aún no está claro si es válido extrapolar estas interpretaciones a datos metagenómicos
Para saber si es posible predecir las tasas de crecimiento a partir de metagenomas, Long y compañía decidieron muestrear comunidades marinas epipelágicas una vez al mes de mayo a septiembre. Después de remover depredadores de bacterias incubaron las comunidades tomando muestras a las 0, 12, 24 y 42 horas para estimar sus densidades poblacionales y secuenciarlas. Armaron 101 genomas de las bacterias y arqueas más importantes del piélago marino, algunas sin parientes cultivables. Las mediciones directas indicaron tasas de crecimiento entre 0.08 y 5.99 células por mL por día. Asimismo, encontraros diferencias explicadas por los grupos taxonómicos y meses del año.
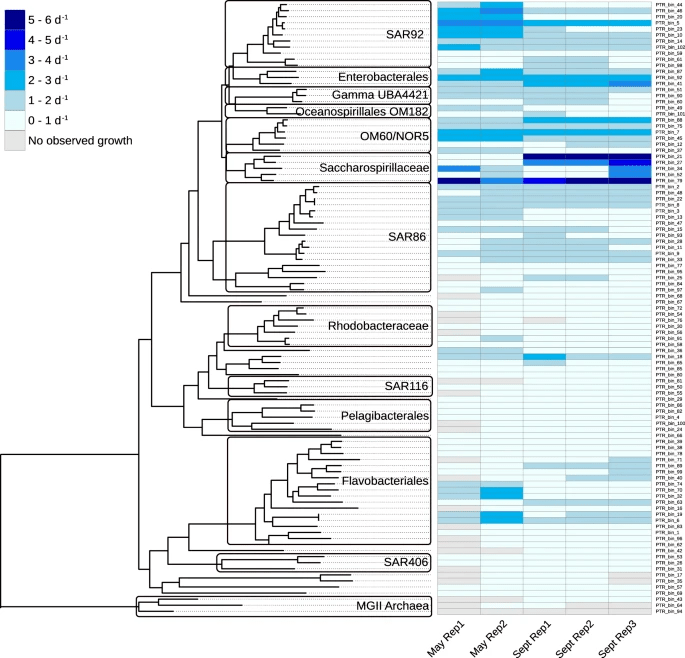
Los autores usaron el CUB para estimar las tasas de crecimiento máximas y encontraron que Oceanospirillales Saccharospirillaceae y Vibrionaceae tuvieron la mayor tasa mientras que Betaproteobacteria, Pelagibacterales y Actinobacteria tuvieron las tasas más bajas. Aunque las tasas de crecimiento real fueron más bajas para 74 genomas, la correlación entre tasas predicha y observada fue de r=0.57 y resultó ser estadísticamente significativa.
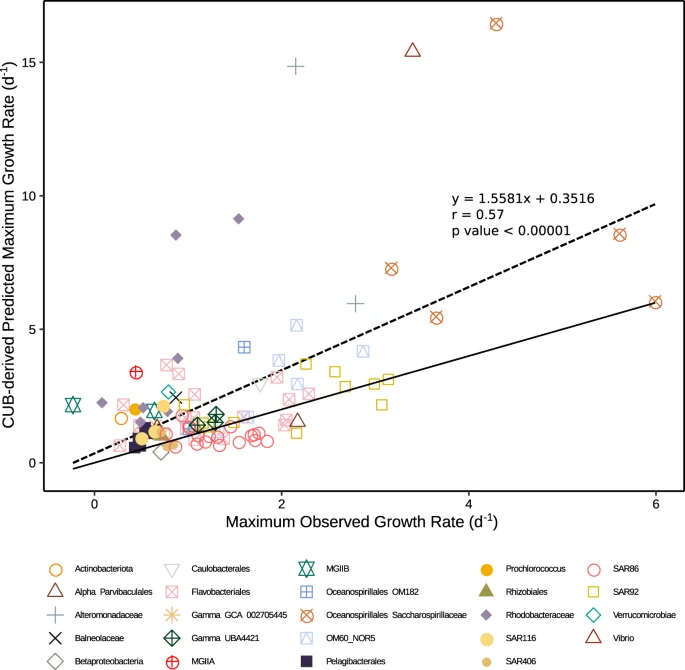
Long y compañía usaron tres métodos para estimar las tasas de crecimiento usando el PTR (iRep, GRiD y DEMIC) y encontraron que al comparar los diferentes valores arrojados estos tuvieron correlaciones débiles o negativas. Al analizar los datos taxón por taxón, encontraron que algunas Gammaproteobacterias tuvieron altas correlaciones entre la tasa estimada y observada. Además las predicciones para Oceanospirillales Saccharospirillaceae tuvieron correlaciones positivas. Además, los índices basados en PTR predijeron crecimiento en genomas que no presentaron crecimiento medible en el laboratorio.
Con respecto a los organismos no cultivables, encontraron un crecimiento mayor al esperado para Pelagibacterales y SAR116 y un crecimiento menor para MGII. Long y compañía concluyen señalando que mientras que el CUB puede proveer estimadores razonables del crecimiento máximo bacteriano, se podría mejorar usando datos transciptómicos y no solamente las secuencias codificantes para proteínas ribosomales. Asimismo, lamentan que el PTR no haya podido generar predicciones aceptables para la mayoría de los organismos. Este trabajo nos ayuda a conocer los métodos más efectivos en la actualidad para estimar tasas de crecimiento y tenerlos a la mano para análisis próximos.
Long, A.M., Hou, S., Ignacio-Espinoza, J.C. et al. Benchmarking microbial growth rate predictions from metagenomes. ISME J (2020). https://doi.org/10.1038/s41396-020-00773-1