Diversidad estructural y evolución de CshA y CshB dos adhesinas presentes en bacterias Gram-positivas y Gram-negativas
Las bacterias han desarrollado múltiples estrategias para colonizar superficies y establecer interacciones con su entorno, entre ellas, destacan las adhesinas fibrilares, proteínas extracelulares alargadas que permiten la unión específica a superficies bióticas (por ejemplo otras bacterias u organismos) o abióticas (como el suelo). Estas adhesinas son clave tanto en interacciones del tipo mutualistas como patogénicas, y su diversidad estructural y funcional representa un reto para su identificación y caracterización.
El estudio presentado por Barringer y colaboradores (2023), se centra en las adhesinas CshA y CshB de Streptococcus gordonii, una bacteria capaz de unirse a fibronectina (Fn), un componente ubicuo de la matriz extracelular. CshA es una adhesina previamente caracterizada, presenta una arquitectura (arreglo estructural) modular que incluye una región N-terminal no repetitiva compuesta por tres dominios (NR1, NR2 y NR3), una región central con múltiples repeticiones y un motivo C-terminal de anclaje a la pared celular de la bacteria (Figura 1A), capáz de unirse a un dominio de fibronectina mediante un mecanismo de reconocimiento y unión, o como lo denominan los autores, tipo “catch-clamp” (Figura 1B), donde el dominio NR1 inicialmente atrapa la fibronectina y el NR2 consolida la unión de forma más estable. El dominio NR2 de CshA forma un núcleo con plegamientos tipo β sándwich, con una hendidura superficial cargada negativamente que constituye el sitio de unión a Fn (Figura 1C).

Figura 1. Estructura y función de CshA. (A) Arquitectura de dominios de CshA, la cual comprende, desde el extremo N-terminal hasta el C-terminal, un péptido señal “FSIRK”, los dominios no repetitivos NR1, NR2 y NR3, 17 dominios repetitivos, y un motivo de anclaje LPxTG. Se indican los límites de los dominios en los extremos N- y C-terminales (las posiciones de secuencia están designadas como “N” y “C”). (B) Ilustración del mecanismo de unión a fibronectina (Fn) tipo “catch-clamp” mediado por los dominios NR1 y NR2 de CshA. La Fn se representa en verde y los dominios de CshA en morado. Se desconoce cuáles dominios específicos de Fn son reconocidos por CshA, por lo que esta ilustración representa una versión simplificada. (C) Representación en cintas (izquierda) y mapa de potencial electrostático de la superficie (derecha; rojo, negativo; azul, positivo; blanco, neutro) del dominio CshA_NR2. La barra de escala electrostática de Poisson-Boltzmann indica un potencial de ±5 kT/e (de rojo a azul, respectivamente). El sitio de unión a Fn en CshA_NR2 se encuentra indicado con un recuadro punteado. Figura tomada de Barringer et al., 2023.
CshB, por su parte, es un parálogo de CshA que comparte aproximadamente un 70% de identidad en secuencia con CshA, pero su dominio de unión (adhesión) NR2 presenta diferencias importantes en la composición electrostática del dominio de unión (Tabla 1). Mediante cristalografía de rayos X, en este trabajo se resolvió la estructura de CshB_NR2 (el dominio de adhesión celular), y se encontró que a pesar de que CshB_NR2 conserva la arquitectura global del dominio de CshA, su cavidad presenta una carga neta menos negativa debido a sustituciones específicas de aminoácidos. Esta diferencia se traduce en una afinidad ∼1.7 veces menor por la fibronectina, como se demostró mediante experimentos de termofluorescencia (MST) (Figura 2). Estos resultados sugieren que CshB podría estar en un proceso de neofuncionalización, en el cual una copia génica duplicada adquiere nuevas funciones gracias a la acumulación de mutaciones que hasta el momento no cambian del todo su función.
Tabla 1. Sustituciones de aminoácidos importantes en CshB_NR2 y sus implicaciones funcionales.
| Posición en CshA_NR2 | Residuo en CshA_NR2 | Residuo en CshB_NR2 | Cambio en la carga | Implicación funcional |
| 300 | Aspartato (D) | Tirosina (Y) | Negativa → Neutra | Disminuye la atracción electrostática con fibronectina |
| 337 | Aspartato (D) | Metionina (M) | Negativa → Hidrofóbica | Reduce la polaridad del sitio de unión |
| 338 | Aspartato (D) | Asparagina (N) | Negativa → Neutra | Disminuye la carga negativa y afecta la especificidad |
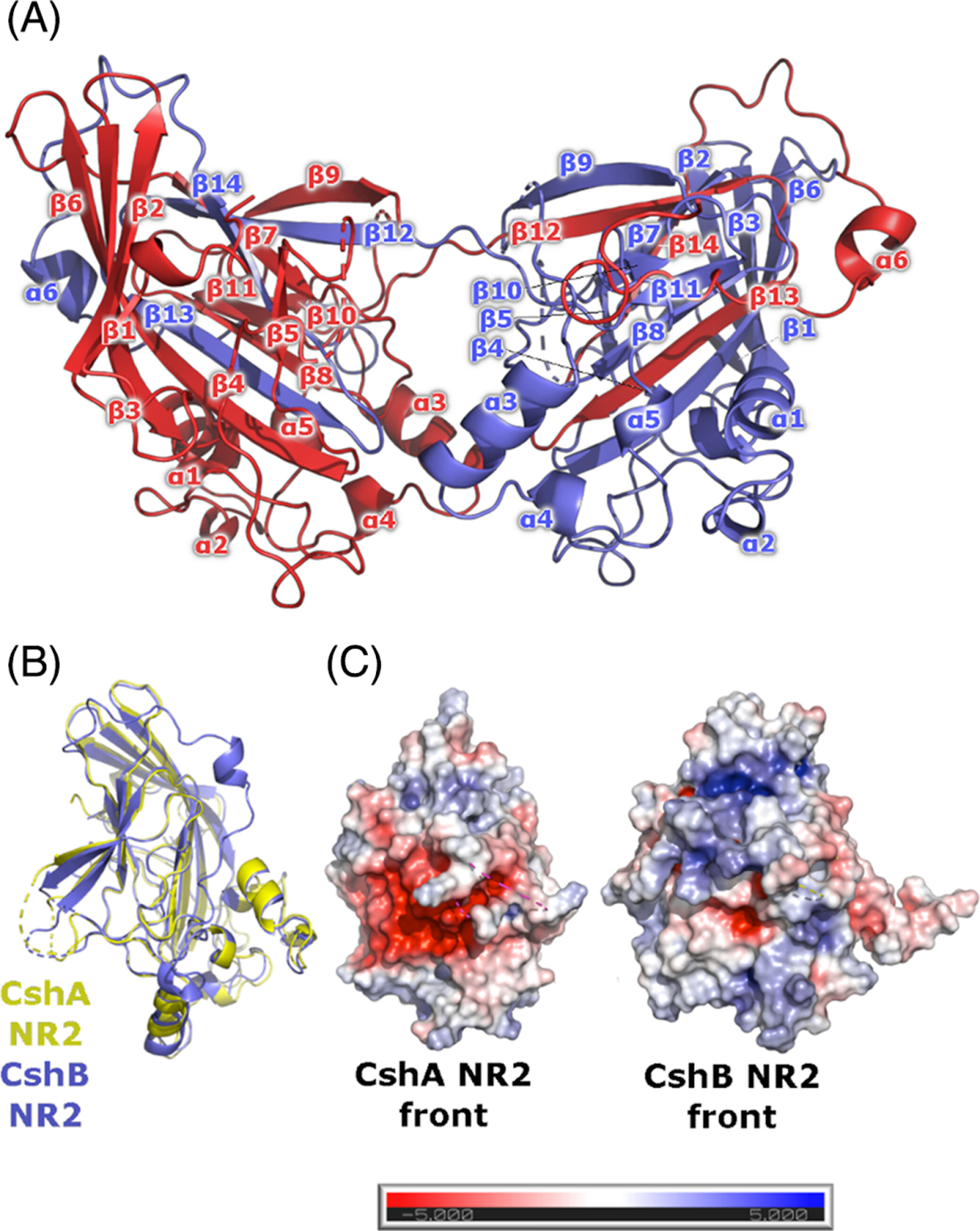
Figura 2. Estructura cristalina de CshB_NR2. (A) Estructura del homodímero de CshB_NR2 con intercambio de cadenas (“strand-swapped”). Las cadenas individuales del monómero están coloreadas diferencialmente (rojo y azul). (B) Superposición de las estructuras del monómero de CshA_NR2 y CshB_NR2. CshA_NR2 se muestra en amarillo, CshB_NR2 en azul. (C) Potenciales electrostáticos superficiales de CshA_NR2 y CshB_NR2 en su forma monomérica. La barra de escala electrostática de Poisson-Boltzmann indica potenciales de ± 5 kT/e (de rojo a azul, respectivamente). Figura tomada de Barringer et al., 2023.
Uno de los hallazgos más relevantes de este trabajo es que señalan la dificultad inherente para identificar adhesinas tipo CshA/CshB mediante métodos basados únicamente en la secuencia, por la cantidad de dominios asociados. A pesar de compartir una estructura altamente conservada, las adhesinas presentan una enorme variabilidad en su secuencia de aminoácidos, especialmente en las regiones funcionales como el hueco de unión y los bucles. Estas observaciones se confirmaron mediante análisis de conservación de residuos y modelado estructural, y mientras que el núcleo de láminas β está bien conservado, los elementos periféricos, como es el caso de los bucles o asaz que regulan el acceso al sitio de unión, son altamente variables entre las diferentes adhesinas analizadas. Posteriormente los autores buscaron homólogos estructurales mediante herramientas como DALI (un programa bioinformático para la la comparación y análisis de estructuras 3D de proteínas), lo que reveló un conjunto de adhesinas con plegamientos similares, todos con capacidad de unirse a componentes de la matriz extracelular, aunque no necesariamente a la fibronectina. Estas incluyen dominios de adhesinas como SpaP, SspB, GbpC, PrgB y Sgo0707. En todos los casos, se conserva el núcleo estructural tipo sándwich β con una cavidad o hueco cargado negativamente, pero se observan variaciones en la longitud y composición de las asaz de acceso al sitio de unión (Figura 3). Estas modificaciones estructurales parecen modular la especificidad de unión del dominio adhesina, permitiendo una gran plasticidad funcional.

Figura 3. Diagramas de topología de los homólogos estructurales de CshA/B_NR2. Las flechas indican las cadenas β, los rectángulos representan las α hélices y las líneas corresponden a las azas. El núcleo conservado tipo sándwich β de cada pliegue está coloreado en azul oscuro. Los elementos estructurales coloreados en azul claro (con contornos punteados) son aquellos dominios ausentes en comparación con CshA/B_NR2, mientras que los coloreados en rojo son dominios adicionales en relación con CshA/B_NR2. Las cadenas β están numeradas conforme al modelo de CshA_NR2, y los elementos adicionales están etiquetados con números romanos. Los elementos estructurales que conforman las asaz que cubren al hueco de unión al ligando en cada uno de los dominios mostrados, están resaltados con recuadros amarillos. Figura tomada de Barringer et al., 2023.
Para entender la distribución evolutiva de estos dominios, se realizó un análisis bioinformático exhaustivo y se identificaron más de 2000 proteínas bacterianas que contienen dominios similares a CshA/B_NR2, tanto en bacterias Gram-positivas como Gram-negativas. Cerca del 28% de estos candidatos pertenecen a bacterias Gram-negativas, muchas de ellas de ambientes marinos o del suelo. La arquitectura de estas proteínas es extremadamente variable, producto de un proceso evolutivo de “barajeo de dominios” (domain shuffling), en el cual diferentes dominios funcionales se combinan en nuevas configuraciones modulares. Esta diversidad se ejemplifica con claridad en las representaciones de arquitectura proteica en la figura 4, en donde el dominio NR2 aparece asociado a una variedad de dominios repetitivos tipo tallo, así como a dominios de anclaje específicos para cada filo bacteriano.
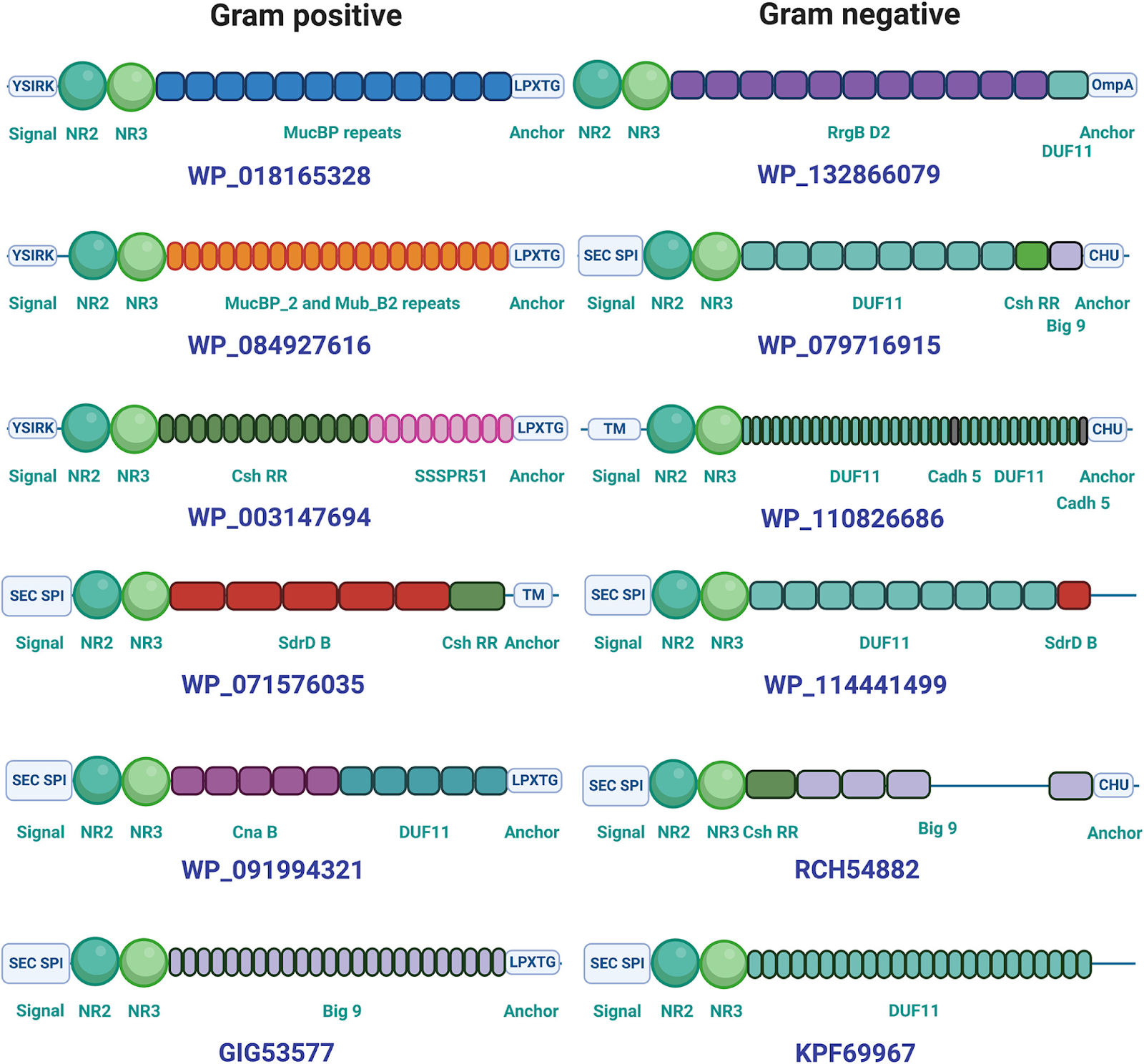
Figura 4. Ejemplos ilustrativos de diversas arquitecturas de dominios en polipéptidos que contienen regiones tipo CshA/B_NR2 identificados en este estudio. Los ejemplos mostrados fueron seleccionados de un conjunto de 2056 secuencias candidatas generadas durante este estudio. Se proporcionan los números de acceso de UniProt correspondientes a las proteínas mostradas. Las diferentes familias de dominios están representadas con distintos colores y etiquetadas con el nombre que se les asigna en la base de datos InterPro. Los dominios no están dibujados a escala. Figura tomada de Barringer et al., 2023.
A pesar de la diversidad mostrada en estos dominios, se puede identificar un patrón recurrente, que es que los dominios NR2 suelen localizarse en el extremo N-terminal de las adhesinas, seguidos de un dominio conservado denominado GEVED (Gly-Glu-Val-Glu-Asp) (anteriormente parte del NR3), y una serie de repeticiones que forman un tallo alargado (Figura 1A y Figura 4). El dominio GEVED ha sido recientemente definido como un nuevo dominio de Pfam (PF20009), con una función aún no totalmente esclarecida pero claramente conservada a lo largo de múltiples linajes bacterianos. El análisis de ubicación relativa y frecuencia de estos dominios refuerza la idea de que las adhesinas evolucionan mediante una estrategia modular, donde el dominio adhesina es colocado en la “punta” de la proteína, pegado al grupo amino terminal, mientras que los tallos repetitivos y los dominios de anclaje permiten la correcta presentación y fijación a la superficie celular. Los autores ilustran en la Figura 5 cómo la reorganización de dominios estructurales (o domain shuffling) ha impulsado la diversificación de adhesinas en bacterias Gram-positivas y Gram-negativas, la figura 5 en el panel(A) muestra la posición relativa de CshA/B con respecto al grupo amino terminal (N-terminus), el panel (B) las combinaciones modulares más comunes entre los dominios de adhesinas, en donde se observa que los dominios tipo CshA/B_NR2, aparecen acoplados a diferentes dominios tipo tallo (stalk), que son estructuras repetidas que alargan la proteína hacia el exterior celular. Esta asociación depende del filo bacteriano, en el caso de Firmicutes, predominan combinaciones con dominios repetidos tipo CshA/B, SSSPR51 y MucBP_2, mientras que en Actinobacteria, se observa mayor uso de dominios tipo CshA_repeat, SpaA y SdrD_B. En Proteobacteria, destacan los dominios repetidos DUF11. En el panel (C) se comparan los motivos de anclaje a la superficie celular y se menciona que todas las adhesinas analizadas presentan un dominio o motivo C-terminal de anclaje, mismo que es coherente con su función extracelular. En bacterias Gram-positivas, este dominio corresponde casi universalmente al motivo sortasa-específico LPXTG, que permite la fijación a la pared celular. En bacterias Gram-negativas, se identificaron cinco tipos de dominios de anclaje, entre ellos los dominios tipo OmpA (comunes en Proteobacteria) y CHU_C (en Bacteroidetes). El panel (D) muestra la media del largo total de las proteínas analizadas por filo bacteriano. En el panel (E) se muestra cómo las diferencias en longitud se correlacionan con variantes estructurales, entre ellos dominios repetidos, en el dominio NR2 así como sus dominios asociados.
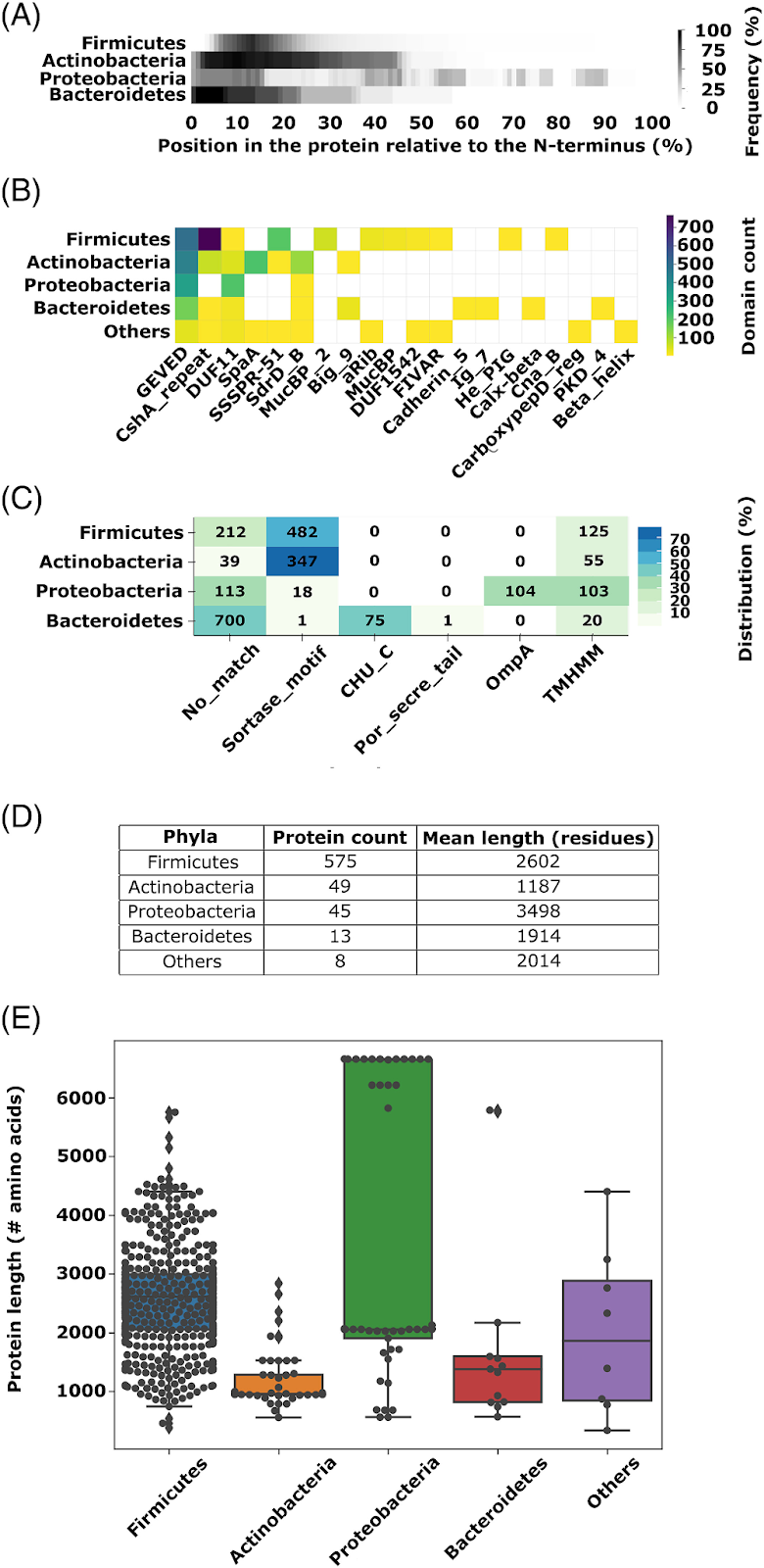
Figura 5. Distribución de los dominios tipo CshA/B_NR2 y de las proteínas que los contienen. (A) Ubicación de los dominios tipo CshA/B_NR2 dentro de polipéptidos de longitud completa, agrupados por filo (690 secuencias analizadas). (B) Longitud de los polipéptidos completos identificados en el conjunto de secuencias obtenidas mediante PSI-BLAST (690 secuencias). (C) Frecuencia de aparición de distintos dominios en proteínas que contienen dominios tipo CshA/B_NR2, clasificada por filo (2056 secuencias). (D) Presencia de péptidos señal y dominios de anclaje en estas proteínas, también agrupada por filo (2056 secuencias). (E) Número total de polipéptidos completos y su longitud promedio, desglosado por filo (690 secuencias). Figura tomada de Barringer et al., 2023.
Referencia:
- Barringer, R., Parnell, A. E., Lafita, A., Monzon, V., Back, C. R., Madej, M., Potempa, J., Nobbs, A. H., Burston, S. G., Bateman, A., & Race, P. R. (2023). Domain shuffling of a highly mutable ligand‐binding fold drives adhesin generation across the bacterial kingdom. Proteins: Structure, Function, and Bioinformatics, 91(8), 1007–1020. https://doi.org/10.1002/prot.26487