Bases de datos para ecosistemas específicos basadas en secuencias completas del gen rRNA 16S
La caracterización de comunidades microbianas a través de la secuenciación masiva de bibliotecas de amplicones del gen ribosomal 16S tiene como limitante a las bases de datos contra las cuales se realiza la anotación. Estas bases de datos sub representan a organismos ambientales y no cultivables, y en ocasiones carecen de rangos taxonómicos completos para los organismos. Además, estos estudios utilizan solamente una fragmento del gen ribosomal, lo cual puede provocar sesgos en la asignación taxonómica, lo cual puede recibir aún más críticas al querer determinar especies bacterianas utilizando definiciones operativas como las secuencias variantes de amplicones (ASV) que recientemente han llamado la atención de la comunidad científica.

En este trabajo, los autores proponen la creación de bases de datos ambientales específicas en donde se generan secuencias completas del gen rRNA 16S y con las cuales se determine su diversidad a través de ASV (FL-ASV, resolución a 1 nucleótido de diferencia). Adicionalmente, nos presentan su pipeline (AutoTax) el cual ofrece una clasificación taxonómica completa de las secuencias utilizando SILVA como referencia y en caso de no poder asignar la clasificación completa, compone nomenclaturas provisionales que pueden permitir la comparación entre distintos conjuntos de datos. Para poner a prueba sus nociones, obtienen DNA y RNA de las comunidades bacterianas de plantas de tratamiento de aguas, generando 1,000,000 de secuencias completas del gen rRNA 16S, los cuales son sujetos al análisis de FL-ASV para obtener la base de datos específica de ese ambiente. Además de esto, secuencian amplicones a partir de las mismas muestras con el objetivo de validar sus resultados y también lo comparan contra conjuntos de datos de otras plantas de tratamiento de aguas.
La base de datos de referencia que generan se compone de 9,521 FL-ASV que son mapeados contra SILVA, encontrando que el 94% de las secuencias tenían un acierto a nivel de género y 74% a nivel de especie. Al mapear amplicones de la región V1-V3 del mismo estudio y otros estudios similares, encontraron que su base de datos logra identificar una mayor proporción de ASVs con alta identidad en comparación con otras bases de datos comúnmente utilizadas (GreenGenes, RDP, etc.). Cuando toman en cuenta a los taxa en baja abundancia (<0.001%) se dan cuenta que hay una disminución en el porcentaje de ASVs que tengan una alta identidad contra las referencias, lo cual es problemático en sistemas con alta diversidad o gran porcentaje de microorganismos transitorios. Para resolver esto, generan una segunda base de datos donde generan OTUs (99% ID) de secuencias completas del gen rRNA 16S (FL-OTUS), obteniendo una mejor resolución en la anotación.
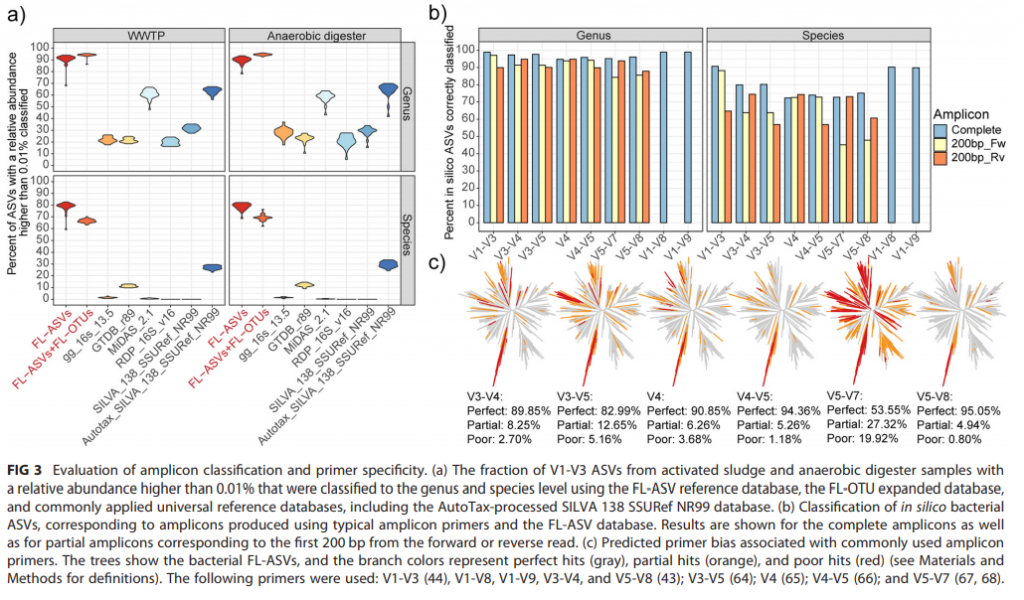
Por otro lado, su sistema de anotación (AutoTax) logró asignar un porcentaje más alto de organismos a nivel de género (89.9%) y especie (78%) comparado contra el resto de bases de datos. Finalmente, evaluaron el desempeño en la clasificación taxonómica de las diferentes regiones hipervariables del gen ribosomal, encontrando que la región V4 ofrece la peor clasificación a nivel de especie, mientras que las secuencias en forward de la región V3 tienen casi el mismo desempeño que las secuencias completas.
En conclusión el trabajo muestra un nuevo método para la clasificación de secuencias en todos los rangos taxonómicos y nos abre las puertas a buscar alternativas para crear nuevas bases de datos específicas para cada ecosistema, lo cual nos puede ayudar a mejores descripciones de las comunidades microbianas.
Dueholm, Morten Simonsen, Kasper Skytte Skytte Andersen, Simon Jon Jon McIlroy, Jannie Munk Kristensen, Erika Yashiro, Soeren Michael Michael Karst, Mads Albertsen, and Per H. Nielsen. «Generation of comprehensive ecosystems-specific reference databases with species-level resolution by high-throughput full-length 16S rRNA gene sequencing and automated taxonomy assignment (AutoTax).» bioRxiv (2020): 672873.