PICRUSt2, una herramienta para la predicción de las funciones en metagenomas.
Una de las estrategias más usadas para la caracterización de las comunidades, es secuenciar genes que están altamente conservados como el 16S rRNA. El cual es un marcador taxonómico y se utiliza mucho para determinar la abundancia relativa de los grupos de bacterias presentes en una comunidad. Sin embargo, las funciones metabólicas de las comunidades no pueden ser inferidas basadas sólo en el uso del 16S rRNA. A pesar de esto, se han desarrollado numerosas herramientas que buscan inferir los perfiles metabólicos de las comunidades partiendo únicamente del uso del gen 16S rRNA. Estas herramientas son: TaxFun, Piphillin, PanFP y PICRUSt1, todos estos algoritmos, infieren las capacidades metabólicas de la comunidad a partir del 16s. De las herramientas antes mencionadas, la más utilizada es PICRUSt1 (publicado en 2013), la cual difiere de las demás porque busca la posición de cada organismo desconocido en un árbol filogenético e infiere estados ocultos que pueden ser empleados. Cabe mencionar que PICRUSt1 presenta algunas limitaciones como: que las secuencias de entrada sean Unidades Operacionales Taxonómicas (OTUs), y que la base de datos de genomas de referencia no se ha actualizado desde el 2013, por esta razón, Douglas y colaboradores decidieron crear una versión mejorada de PICRUSt1, esta versión la llaman PICRUSt2 y acaba de ser liberada en el 2020.
En PICRUSt2, las mejoras que se hicieron tienen que ver con los datos de entrada. Esta nueva versión, permite la entrada de OTUs o ASVs (del inglés Amplicon Sequence Variants), estos últimos tienen una resolución más fina, lo que permite distinguir a los organismos que están más relacionados. El número de genomas de referencia es de 41, 926 los cuales vienen de la base de datos de IMG (del inglés Integrated Microbial Genomes), cabe destacar que muchos de estos genomas son de cepas de la misma especie y poseen un 16S rRNA idéntico. Los OTUs o ASVs que provienen de una muestra se colocan en una filogenia preexistente. El posicionamiento filogenético de PICRUSt2, se basa en la salida de tres herramientas: HMMER para ubicar a los ASVs, EPA-ng que determina el posicionamiento óptimo de estos ASVs en la filogenia de referencia y GAPPA que genera un nuevo árbol filogenético incorporando los reemplazamientos por ASVs. Esto da como resultado un árbol que contiene ambos, los genomas de referencia y los organismos muestreados ambientalmente, los cuales son usados para predecir las familias de genes individuales y el número de copias para cada AVS. Este procedimiento se hace para cada conjunto de datos de manera individual, lo que le permite al usuario diseñar su propia base de datos de acuerdo con sus necesidades, incluyendo aquellas que puedan ser optimizadas para el estudio específico de ciertos ambientes microbianos. Además de proveer la contribución de cada ASV para cada predicción de la función lo que permite tener análisis estadísticos basados en taxonomía (Fig. 1).

Fig. 1 | PICRUSt2 algorithm. a, The PICRUSt2 method consists of phylogenetic placement, hidden-state prediction and sample-wise gene and pathway abundance tabulation. ASV sequences and abundances are taken as input, and gene family and pathway abundances are output. All necessary reference tree and trait databases for the default workflow are included in the PICRUSt2 implementation. b, The default PICRUSt1 pipeline restricted predictions to reference OTUs in the Greengenes database. This requirement resulted in the exclusion of many study sequences across four representative 16S rRNA gene sequencing datasets. PICRUSt2 relaxes this requirement and is agnostic to whether the input sequences are within a reference database or not, which results in almost all of the input ASVs being retained in the final output. c, An increase in the taxonomic diversity in the default PICRUSt2 database is observed compared to PICRUSt1.
Como la base de datos aumenta, se incrementa la diversidad taxonómica de PICRUSt2. Al incrementar la diversidad, las predicciones de las familias de genes reportadas en la base de datos de familias de genes y genomas de la enciclopedia de Kyoto (KEGG), ortólogos (KOs) y números de la comisión de enzimas (EC) también aumenta. Para validar esta herramienta se utilizaron siete conjuntos de datos que se encuentran públicos, entre los que se incluyeron amplicones de los 16S y de secuenciación de metagenomas (MGS) de muestras del microbioma humano y de rizosfera. Y se compararon con los cuatro predictores de función: PICRUSt1, Piphillin, PanFP y Tax4Fun2. Las métricas de correlación representan los rangos de similitud de las abundancias entre los datos predichos y los datos observados. En todas las muestras los datos predichos con PICRUSt2 fueron mejores que para los otros métodos. Por lo que esto podría reflejar una diferencia entre usar los métodos basados en filogenias sobre aquellos que no usan filogenias. En conclusión, las familias de genes se correlacionaron en el número de copia a través de todos los taxa. Las correlaciones fueron muy fuertes dentro y entre los diferentes ambientes. Una aproximación complementaria para validar las predicciones de los metagenomas, es comparar el resultado de las abundancias diferenciales en los metagenomas predichos con los datos de MGS. Del análisis de precisión llevado a cabo para PICRUSt2 junto con las otras herramientas, se observó que PICRUSt2 muestra la medida de precisión y recuperación más alta en comparación con los otros métodos. Además, cuando se compararon estos resultados con las pruebas de abundancia diferencial de los KOs de las MGS se obtuvo un ligero incremento en la precisión. Estos resultados resaltan la dificultad de la predicción funcional, partiendo de los marcadores y de los datos metagenómicos actuales (Fig. 2).
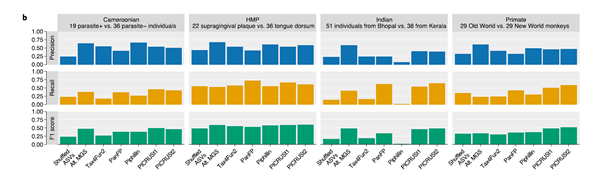
Fig. 2 | PICRUSt2 performance characteristics. Validation of PICRUSt2 KO predictions comparing metagenome prediction performance against gold-standard shotgun MGS. b, Comparison of significantly differentially abundant KOs between predicted metagenomes and MGS. Precision, recall and F1 score are reported for each category compared to the MGS data. Precision corresponds to the proportion of significant KOs for that category also significant in the MGS data. Recall corresponds to the proportion of significant KOs in the MGS data also significant for that category. The F1 score is the harmonic mean of these metrics. The subsets of the four datasets compared are indicated above each panel (the Cameroonian parasite is Entamoeba). Wilcoxon tests were performed on the KO relative abundances after normalizing by the median number of universal single-copy genes per sample. Significance was defined at a false discovery rate <0.05. The Shuffled ASVs category corresponds to PICRUSt2 predictions with ASV labels shuffled per dataset. The Alt. MGS category corresponds to an alternative MGS processing pipeline with reads aligned to the KEGG database rather than the default HUMAnN2 pipeline (Modificado de Douglas et al., 2020).
La base de datos de MetaCyt es una alternativa a KEGG y también se ha usado mucho para estudios de metagenómica funcional. La abundancia de las rutas de MetaCyc son calculadas en PICRUSt2 a través de mapeos de los números de la comisión de enzimas, y de familias de genes en las rutas. Las abundancias en las rutas de MetaCyt son las predicciones que dieron la mejor salida para PICRUSt2. Cabe resaltar que las pruebas de abundancia diferencial de las rutas mostraron una variabilidad alta través de todos los conjuntos de datos. Estos resultados, sugieren que la identificación robusta diferencial de la abundancia de las rutas en los metagenomas es difícil, resaltando el reto de analizar rutas metabólicas generales en las bacterias. Así mismo, se hicieron las predicciones para 41 fenotipos microbianos vinculados a los genomas que ese encuentran en IMG, y estas predicciones pueden ser generadas con PICRUSt2. Estas representan actividades metabólicas de alto nivel como utilización de la glucosa y desnitrificación. Que son anotadas como presentes o ausentes en cada uno de los genomas de referencia.
Así mismo existen dos críticas fuertes a la predicción funcional basada en amplicones. La primera es el sesgo que existe en las predicciones hacia el uso de los genomas de referencia, lo que quiere decir que funciones específicas a cada uno de los ambientes no son fáciles de identificar. Sin embargo, esta limitación empieza a decaer a medida que están disponibles más datos. PICRUSt2 permite al usuario, seleccionar genomas para ser usados en el análisis, para generar predicciones, las cuales proveen de un análisis flexible si se quieren estudiar ambientes particulares. La segunda crítica es que las predicciones basadas en amplicones no pueden proveer la resolución para distinguir la especificidad funcional de las cepas, lo que es una gran limitante para PICRUSt2. Finalmente, se espera que PICRUSt2 permita la identificación de las funciones principales y las implicaciones ecológicas de los microorganismos que componen a la comunidad basados únicamente en los amplicones del gen 16S rRNA.
Referencia
Douglas, G.M., Maffei, V.J., Zaneveld, J.R. et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 38, 685–688 (2020).