Evaluación de la secuenciación del gen 16S rRNA para análisis de microbiomas a nivel de cepas y especies
Con la secuenciación en masa de amplicones de 16S la norma ha sido generar clusters basados en similaridad para generar las unidades taxonómicas de operación. Con los avances de la secuenciación se han aumentado el largo de los fragmentos con los se analizan las comunidades bacterianas. Una de las más recientes es la secuenciación de fragmentos completos de 16S donde se podrá apreciar y hacer más fina la asignación taxonómica de las bacterias. El uso de las regiones hipervariables del gen 16S ribosomal aunque útil no está exento de algunos problemas o deficiencias que pueden o no mejorarse con la secuenciación de fragmentos más grandes del mismo.
Los nuevos métodos de secuenciación circular mencionan que tienen la capacidad para obtener la secuencia completa de 16S y discriminar entre secuencias con 1 solo nucleótido de diferencia. Los autores de este trabajo utilizan este sistema para comparar la asignación taxonómica con respecto a otras técnicas como la secuenciación completa del genoma de algunas bacterias ya caracterizadas tanto a nivel in vitro e in silico.
la primera parte fue el análisis de secuencias de 16S no redundantes >1% de diferencia y que fueron cortadas según los fragmentos generados por los amplicones más comúnmente utilizados para este tipo de análisis y se comparó la asignación taxonómica generada con diferentes fragmentos y la que se obtiene con la secuencia completa.
Se encontró que la mayor cantidad de secuencias asignadas correctamente fue al utilizar los fragmentos completos pero que las subregiones representaron de manera eficiente la asignación taxonómica en algunas lo suficiente para la asignación de especie. También se vio que la asignación y eficiencia difiere dependiendo del fragmento utilizado. la región V6-v9 resultó ser eficiente para la asignación de Clostridium y Staphylococcus, la V3-V5 para Klebsiella y V1-V3 para Escherichia/Shigella. La que peor rendimiento tuvo fue la región V4.

Para comparar si la eficiencia calculada anteriormente se reflejaba secuenciaron de manera completa fragmentos de 16S de diferentes cepas de E.coli por medio de PacBio y Illumina Hiseq y compararon para ver si la tasa de errores era debido a la secuenciación o era un reflejo de los poliformismos de las diferentes cepas. Al ver que con secuencias largas pueden identificar polimorfismos del mismo genoma demuestra que no es válido asumir que secuencias que varían en uno o pocos nucleótidos representan diferentes taxas.
Para verificar la eficiencia de la secuenciación completa calcularon la abundancia de Bacteroides a nivel de género y a nivel completo por mWGS como V1-V3 Illumina y V1-V9 PacBio la secuenciación produjo resultados comparables. Ambos acercamientos identificaron a los individuos con baja proporción de Bacteroidetes de los dos con alta.
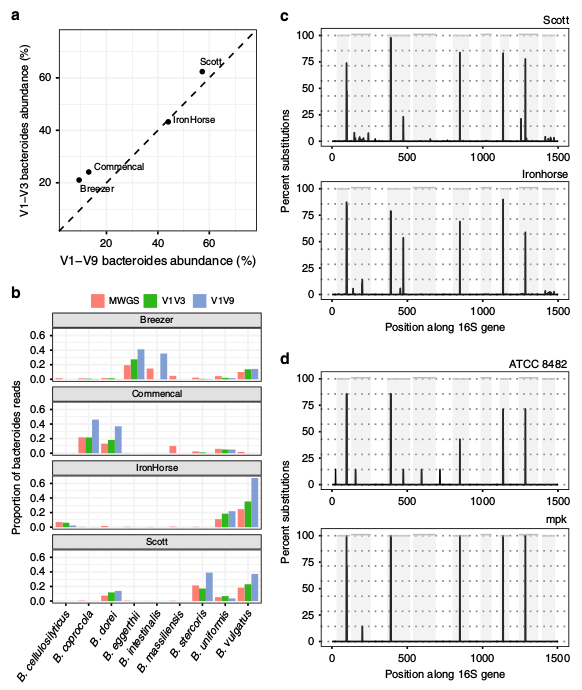
Ya que es posible resolver entre variantes intra genómicas que aparecen en el mismo taxa, establecieron que perfiles pueden ser utilizados de manera rutinaria para distinguir entre cepas de la misma especie. Ellos cultivaron 381 taxas del microbioma de personas sanas y generaron de ellas secuenciaciones completas del 16S para identificar sustituciones nucleotídicas características de copias intragénicas de 16S. De estas se identificaron 58 especies, mientras que a 99% de similitud se agrupan 61 OTUs (con entre 61 y 73 aislamientos a cada OTU). En total 349 de 381 aislados (54 de 61 OTU). Esto indica la presencia de polimorfismos y se identificaron 205 perfiles de SNP únicos al tener en cuenta un posible error de secuenciación

Johnson, J.S., Spakowicz, D.J., Hong, B. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun10, 5029 (2019). https://doi.org/10.1038/s41467-019-13036-1