Búsqueda de ortólogos funcionales por medio de modelos ocultos de Markov
Las bases de datos de la Enciclopedia de Kyoto de Genes y Genomas (KEGG) son ampliamente usadas para analizar genomas y metagenomas bacterianos por su información detallada y la posibilidad de analizar rutas metabólicas visualmente. En el portal de internet de esta institución, existen servicios de anotación de secuencias como son el BlastKoala, GhostKoala y KAAS, cada uno con sus objetivos, métodos y supuestos particulares. En este trabajo se describe una nueva herramienta de anotación de secuencias llamada KofamKOALA que usa perfiles de proteínas generados con modelos ocultos de Markov de alineamientos múltiples de ortólogos funcionales que promete ser particularmente veloz.
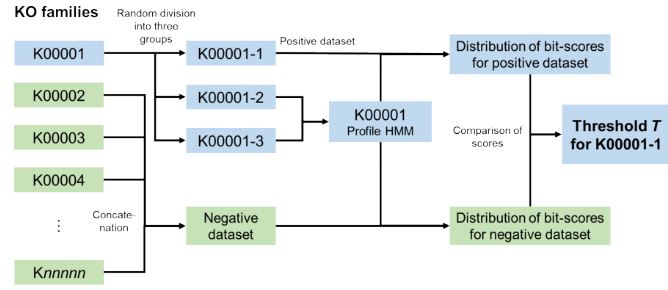
Ya que los perfiles de alineamientos de proteínas permiten encontrar secuencias homólogas lejanas, es necesario establecer valores de corte para evitar falsos positivos cuando se usan para buscar secuencias en bases de datos. Por esta razón, Takuya Aramaki y compañía establecieron valores de corte por medio de una estrategia de validación cruzada que toma un subconjunto de las secuencias semilla para generar un perfil de proteínas y usa los otros grupos para calcular la distribución de bitscores que se pueden encontrar en las secuencias verdaderas-positivas. De esta forma, se establece un valor de corte de bitscore que ayuda a decidir si un acierto en la búsqueda de secuencias es un verdadero-positivo.
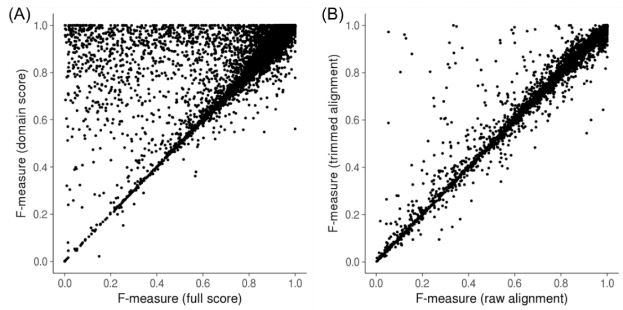
Como el puntaje de alineamiento depende de la longitud de las secuencias y cómo pueden haber homólogos funcionales lejanos, los autores revisaron el efecto de tomar en cuenta el alineamiento completo o solo el dominio mejor alineado y de quitar secuencias muy divergentes del alineamiento múltiple. Dependiendo de los resultados de esta prueba, los autores especificaron en Kofam qué bitscore debería revisar y cotejar con su valor de corte correspondiente, el del alineamiento completo o el de la proteína completa. Al comparar el desempeño de KofamKOALA con otras herramientas del KEGG, se encontró que de hecho es la herramienta más veloz, aunque en términos de sensibilidad es superada por BlastKoala y GhostKoala, dependiendo de los organismos que se incorporen al análisis.
Los autores señalan que esta herramienta se puede usar idealmente con genomas, ya que si se buscan secuencias incompletas como las que se pueden encontrar en bases de datos metagenómicas, estas pueden afectar el puntaje de alineamiento y por ende hacer inservible el valor de corte previamente establecido para cada perfil. Este trabajo nos enseña cómo es posible automatizar el establecimiento de valores de corte cuando es necesario llevar a cabo búsquedas con un gran número de modelos ocultos de Markov sobre una base de datos.
Aramaki, T., Blanc-Mathieu, R., Endo, H., Ohkubo, K., Kanehisa, M., Goto, S., & Ogata, H. (2020). KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics, 36(7), 2251-2252.