PhyloPhlAn 3 permite hacer comparaciones filogenómicas a diferentes escalas
En este trabajo, se reportan los avances en la nueva versión de PhyloPhlAn, con la cual es posible comparar genomas estrechamente emparentados o bien miles de genomas de phyla distintos de forma eficiente. Esto se logra gracias a la incorporación de información como los 87,173 genomas de referencia y los 154,723 genomas ensamblados a partir de metagenomas de GenBank, y las más de 57 millones de familias de proteínas de UniRef90. Asnicar y compañía reportan que para comparar genomas emparentados, este programa usa un dataset de más de 18,000 conjuntos de genes pre-seleccionados de UniRef, mientras que para comparar genomas lejanos, usa los 400 marcadores moleculares más conservados. Este software puede incorporar distintos programas a su serie de pasos para adaptarse a las preferencias del usuario.
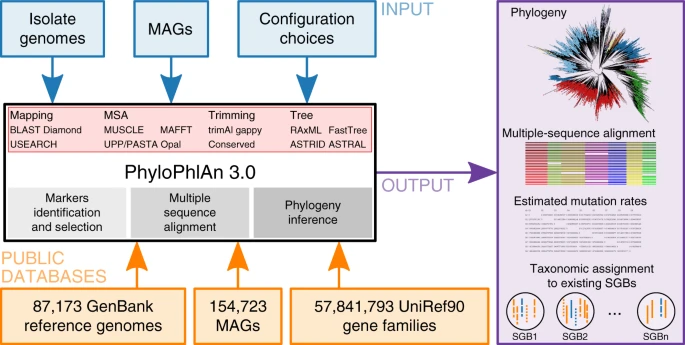
Este programa tiene cuatro pasos principales: identificación de marcadores moleculares, refinamiento del alineamiento múltiple, concatenación de alineamientos y la construcción de la filogenia. Los autores mencionan que una de las utilidades de este programa es la clasificación de genomas ensamblados con lecturas metagenómicas (MAGs, por sus siglas en ingles) y muestran un ejemplo con datos de metagenomas intestinales. De entre los 369 MAGs analizados, PhyloPhlAn logró clasificar 352 de ellos a nivel de especie, aunque el 39% carece de etiqueta taxonómica. Otros 17 genomas correspondieron a especies candidato que no se habían observado antes
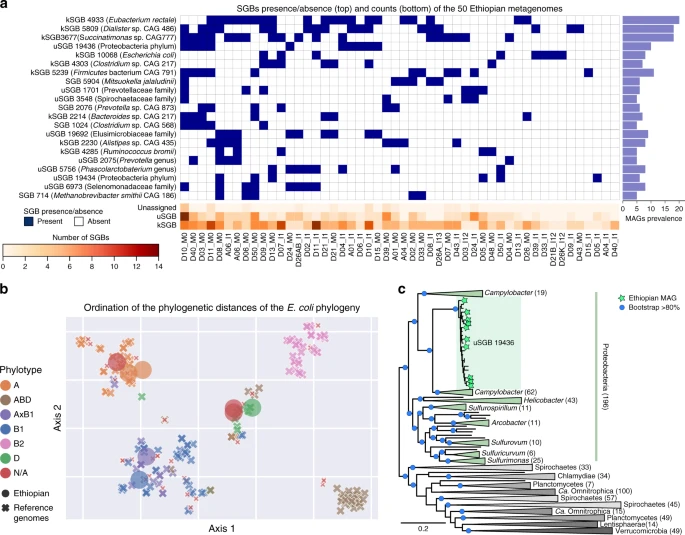
Para demostrar la capacidad de PhyloPhlAn 3 para llevar a cabo comparaciones a gran escala, los autores analizaron un dataset de 17,672 genomas representativos no redundantes. Encontraron una dominancia de proteobacterias en muestras no humanas y de actinobacterias en muestras humanas. La construcción de este árbol tardó 10 días usando 100 núcleos con el programa IQ-TREE. El alineamiento de 4,522 aminoácidos incluyó genomas de bacterias y arqueas, varios siendo MAGs.
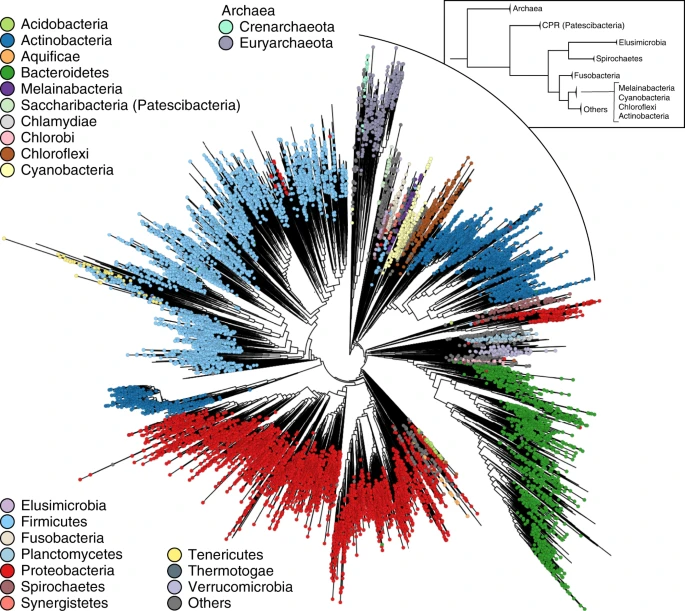
Esta herramienta es una poderosa alternativa para llevar a cabo análisis filogenómicos y clasificar MAGs que podríamos encontrar en comunidades ambientales.
Asnicar, F., Thomas, A.M., Beghini, F. et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0.Nat Commun11, 2500 (2020). https://doi.org/10.1038/s41467-020-16366-7